












Reimagine Clinical Research
Connect Stakeholders
Bring everyone together and keep them connected to the information they need to accelerate clinical trial outcomes. Learn More >
Protect Trial Participants
We’ve been patient-centric since the beginning, and our research review services are unmatched in the category. Learn More >
Deliver Innovation
We don’t rest in our mission to make clinical research safer, smarter, and faster for patients, sites, sponsors and CROs. Learn More >

Proven Clinical Research Services and Technology

#1 in Trusted Research
Review Services

#1 in Connected Site and
Sponsor Technology

Reimagining the Clinical
Research Experience
Top Clinical Research Sponsors, CROs, and Sites Partner with Advarra to Accelerate Their Clinical Research
institutions, hospitals, health systems, and academic medical centers trust Advarra.
investigators utilize Advarra trial technology solutions to support their research.
open protocols overseen by Advarra annually
of NCI-designated Cancer Centers use Advarra technology.
Bring Your Clinical Trial Together
Advarra’s IRB and IBC reviews and Gene Therapy Ready site network helped reduce study startup timelines and delivered trial results to IQVIA quicker than expected.

At Inova Health System, the research team improved regulatory efficiency by 20% without the need for extra regulatory staff by adopting Advarra’s eRegulatory Management System.
Using Advarra Insights, The University of Wisconsin Carbone Cancer Center was able to enhance their data analytics and visualization capabilities.

Advarra served as the institutional review board (IRB) of record for the phase I trial for the first-in-human dose-escalation study of an mRNA Vaccine Against SARS-CoV-2.
Longboat:
The Reimagined
Clinical Trial Experience
Longboat is the single hub to centralize, connect, and simplify your clinical trials
Create a central hub for a study to reduce burden and increase visibility
Modern, simple technology that sites want to use
Faster startup and improved regulatory process efficiency
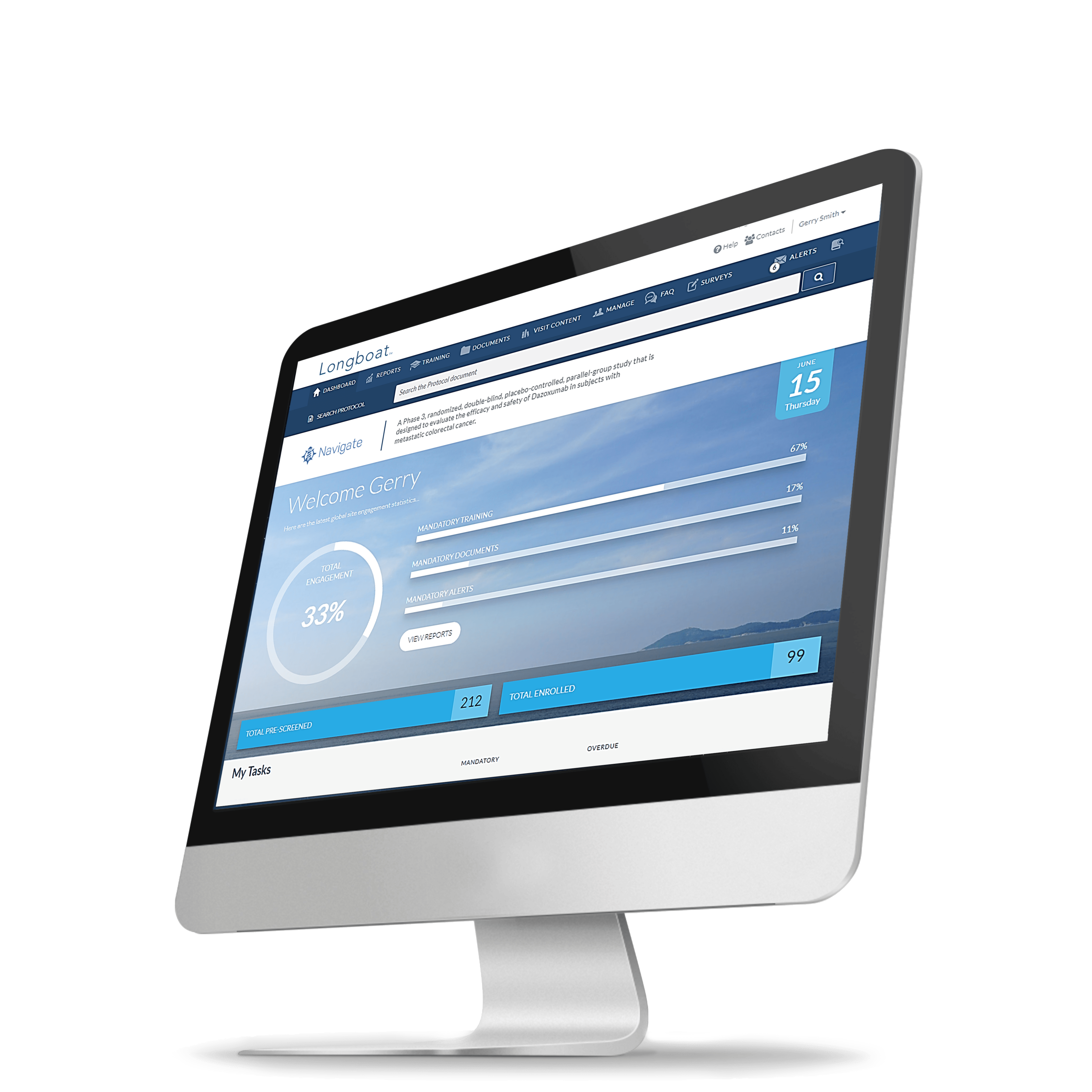

The 2023 Study
Activation Report
Hear from clinical researchers striving to get life-saving therapies into the market.
Read about
- How a proliferation of technology and process is creating delays and adding administrative burdens that stall clinical research progress.
- What sponsors and CROs are doing to reduce site feasibility burdens.
- Which technology improvements are impacting study activation strategies.
Are You Ready to Bring People Together for Better, Faster Trial Outcomes?
Subscribe to our monthly email
Receive updates monthly about webinars for CEUs, white papers, podcasts, and more.
Advancing Clinical Research: Safer, Smarter, Faster™
Copyright © 2024 Advarra. All Rights Reserved.